What if we viewed lived experience as an asset? As an asset of professional, economic,…

My Stay At The Neuromuscular Complex Care Centre

Many of you will know, in November 2016 it was confirmed that I have some form of neuromuscular disease by the fantastic team at the National Hospital for Neurology and Neurosurgery (NHNN) and would be embarking on a long process to try and find a diagnosis. Dr Ros Quinlivan doubts my Ehlers-Danlos Syndrome diagnosis, rather than having ligament laxity, it is my weak (and some undeveloped) muscles that cause my joints to be lax. The neuromuscular disease is far more likely to cause the wide ranging, degenerative complications than EDS is, as everyone in my team have for a long time felt that EDS just didn’t ‘fit’. It could be possible to have both EDS and a neuromuscular disease, but it’s unlikely – that’d involve winning the genetic lottery twice, I’m not sure I’m that lucky (or unlucky). We have a long diagnostic process to plough through, it is very likely I have a rare or even unique genetic condition; especially since nothing runs in my family.
Since November when I saw Dr Quinlivan, I’ve been waiting for an admission for testing; this admission occurred this past week, Monday 10th – Friday 14th July. Whilst no diagnosis has been reached – it’ll be a long process – the week has been productive. I thought I’d give a recap/update of the admission for those who are interested.
Bloods:
I had bloods taken for the 100,000 Genomes Project, to try and find the diagnosis.
I also had some metabolic bloods too.
Respiratory:
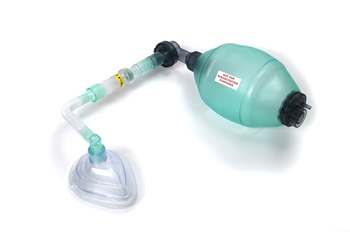
I had some lung function tests, which showed my respiratory muscles are weak and my lung capacity is low and the force at which I can expel air is poor. I’m breathing in my upper chest and air isn’t reaching the bases of my lungs. My cough is weak too. I’ve recently (pre-admission) been started on twice daily saline nebulisers to loosen up the secretions that build up whilst I am asleep, which combined with my weak cough are hard to clear. I’ve been given a Nippy Lung Volume Recruitment bag and mask to help the air reach the bases of my lungs. Here is a picture. Basically, I press the mask above my face, someone squeezes the bag as I take a breath in, then we air stack, which means I take little breaths as they squeeze the bag to force more air into my lungs to get enough air to reach the bases. I then remove the mask and breathe out or cough. This is to help air reach the bases of my lungs, forcing air in as I cannot naturally breathe in enough to fill my lungs, and I can then use that to mobilise secretions and/or to produce a better, more effective cough. That, combined with the twice daily nebuliser, should prevent chest infections by giving me a better clearance of my chest. I’ve also got to keep recording my cough volume to ensure things aren’t deteriorating.
I had a sleep study which showed my blood carbon dioxide level (PaCO2) does rise in the night but fortunately no big oxygen desaturation and the CO2 rise is not alarming at this point. I don’t meet the requirement for needing non-invasive ventilation (thank goodness), so we can just keep an eye on things for now.
Metabolic:
I was reviewed by Dr Robin Lachmann, who said no metabolic diseases stood out but that a Mitochondrial Disease was likely.
Neurogastroenterology:
I’m already under Professor Qasim Aziz and have been since 2010 but they wanted me to see Dr Anton Emmanuel, the neurogastroenterologist based at UCL hospitals. He didn’t have much to add, other than to restrict my fluid intake as it’s making the situation worse with my gut. It’ll be hard as I feel desperately thirsty 24/7 but if it’ll help things then I will do it, as it’s worth a try. He didn’t have anything else to add, but he did educate his students on my “end stage Intestinal Failure”. He was lovely and I’m glad to have met him.
Physiotherapy:
I saw the physiotherapist who, in addition to the respiratory interventions, gave me some resistance bands to try and maintain my arm strength or at least slow its decline. We won’t build muscle but maintaining the strength I have now is the ultimate goal. She was really lovely, and did pick up on the Hypermobility in my joints.
Cardiology:
My heart is okay, my ECG is not too bad and echocardiogram shows nothing of much concern, which is fantastic.
Sadly, despite the team there trying hard, my muscle biopsy couldn’t be organised whilst I was inpatient. They did their best. I just hope it won’t be too long before we can organise it, as it will potentially lead to a diagnosis or if not, narrow down the possibilities. So the muscle biopsy will be a waiting game, but the Genomes bloods have been sent off so they’ll be sequencing it and looking at that, but that’s a very long process – like finding a single spelling mistake in a library full of books. It could take years, so that won’t be a quick process.
I feel very positive. We’ve got management strategies in place for my respiratory problems, I’ve had some tests, things are moving forwards and hopefully the muscle biopsy will be organised soon. The stay was not bad, the Neuromuscular Complex Care Centre (NMCCC) staff are fantastic and despite some teething problems with TPN and things the admission went smoothly. I even managed to leave the ward twice to get out with mum, which was nice, a luxury I’m not afforded in most admissions as it takes so much time and manpower to get me out of bed and ready and to time in between IVs and everything, most wards can’t allow me or facilitate me leaving but this admission we went out twice in the evening to escape the ward for a bit. I’ve met some lovely people too. So it was definitely a positive admission, and was step one on the road to a (hopeful) diagnosis.
I am now home and reunited with my gorgeous companion and Assistance Dog Molly.
Lucy x


Related Posts



so glad your stay went well. i hope you get some questions answered. it's not easy being rare,as how do you treat an illness when you don't know what it is. sending you all the best wishes in the world. from one rarity to another ��