What if we viewed lived experience as an asset? As an asset of professional, economic,…

Rare Disease Day 2017 – Do You Care About Rare?

According to Rare Disease UK, rare diseases affect 1 in 17 of us at some point in our lifetime, which translates to 7% of the population being affected by a rare disease during their life. This equates to 3.5 million people in the UK, or 30 million across Europe. With as many as 6,000-8,000 rare diseases, it’s high time doctors accepted that ‘rare’ is more common than we think. The individuals with these conditions can be as few as a handful or even just one person, or affect as many as 20-30,000 people. 80% of rare diseases have a genetic component and 75% of rare diseases affect children. Rare diseases can be chronic and even life-threatening and life-limiting, and include conditions right from super-rare conditions like Field’s Condition, only known to affect and named after Catherine and Kirstie Fields, ranging to conditions such as Fibrodysplasia Ossificans Progressiva (FOP), with 700 cases reported worldwide, as well as more prevalent conditions such as Cystic Fibrosis, affecting approximately 8,000 people in the U.K.
So that’s the statistics; rare diseases aren’t as rare as you think! However, when you break the conditions down, rare diseases can affect tens of thousands, or just one or two people.

I have a rare disease. You all know I was diagnosed with Hypermobile Ehlers-Danlos Syndrome when I was 15, and EDS Hypermobility is not a rare condition. Other forms of EDS, like Vascular, Kyphoscoliotic and Arthrochalasic EDS are. So Hypermobile EDS didn’t make me rare, despite the misconception in the EDS community about it being so. Still, I have the zebra pattern stuff that is the mascot for rare disease, and has been adopted as the Ehlers-Danlos mascot. The saying goes in medical school “When you hear hoofbeats, think horses, not zebras.”
Doctors are taught to think of common conditions (horses) rather than rare conditions (zebras), they are taught to think of the simplest and most common causes and conditions, rather than matching symptoms to rare diseases. Also, zebras stripes are never the same, much like our fingerprint, and this is said to represent how no two EDS patients are the same, just like a zebras stripe pattern is unique to them.
However, my Ehlers-Danlos Syndrome diagnosis was questioned last year. I could not have EDS at all, or I have EDS as well as another condition, which explains the severity, complexity and progressive nature of my condition, and why it affects so many organs and systems. The neuromuscular specialist told me I likely have some rare genetic neuromuscular (muscle-wasting) disorder, or a rare form of Mitochondrial Disease. Finally everything made sense, why I was born with health problems, I was born with a dislocated hip, had severe silent reflux, a floppy airway that would stop me breathing if I was laid flat and severe constipation. I had problems throughout my childhood, many problems, septic arthritis from the undiagnosed dislocated hip, flat feet, constipation, tummy pain, weakness of my muscles, Hypermobile joints, joint deformities, eye problems and more; but no one connected the dots, I saw different people for different problems and no one thought it was odd. I started physiotherapy age 11, and my physio couldn’t understand how I was very physically active, horse riding most days, working at the yard mucking out, sweeping, carrying heavy objects like hay nets, hay bales, feed bags and water buckets, grooming the horses, doing lots of walking, pushing wheelbarrows and the actual riding itself. I was doing this almost every day. Yet, my muscles were weak, some completely undeveloped and they couldn’t support my joints at all. It made sense why I was falling a lot from my legs giving way and struggling to write, to walk, I couldn’t run and why I was struggling to keep up with my active life, needing more and more help. I struggled to write, I had to go up and down stairs on my bottom, I couldn’t get up off the floor, and I needed help to wash my hair. Not only were my muscles weak, but despite religiously doing my physiotherapy exercises, my muscles were getting weaker and not stronger. So despite how much I was doing and how hard I worked on my physio, my muscles were getting weaker still. It didn’t make sense and my physiotherapist regularly took my mum aside and said he knew something was wrong, but he didn’t know what.

In January 2008, age 14, I became wheelchair dependent. I got my diagnosis of EDS a few days after my 15th birthday in the October. I continued to deteriorate though. I became bed bound in 2009. My gut failed and I started tube feeds in August 2009, was diagnosed with Chronic Intestinal Pseudo Obstruction and POTS in 2010, started having to catheterise to empty my bladder in 2010, went onto TPN (intravenous nutrition) in May 2011 after nearly dying, having a heart attack from my body digesting my heart muscle and my bone marrow starting to fail. In 2012 I had my Ileostomy formed, in 2013 I had a permanent catheter (suprapubic catheter) placed, in 2014 I had a Urostomy (Ileal Conduit) formed and as such no longer had the suprapubic. I now have a collection of secondary conditions and complications: complete failure of my digestive system (type 3 Intestinal Failure), neurogenic bladder failure, severe problems with my Autonomic Nervous System (Autonomic Neuropathy and POTS), as well as problems with my bones (Osteoporosis and early-onset arthritis), spine (Kyphoscoliosis, spinal degeneration and others), lungs (Restrictive Lung Disease and gas exchange abnormalities), metabolic and endocrine problems, immune system dysfunction and problems with my kidneys. My muscles are progressively atrophying too, they’re just wasting away.
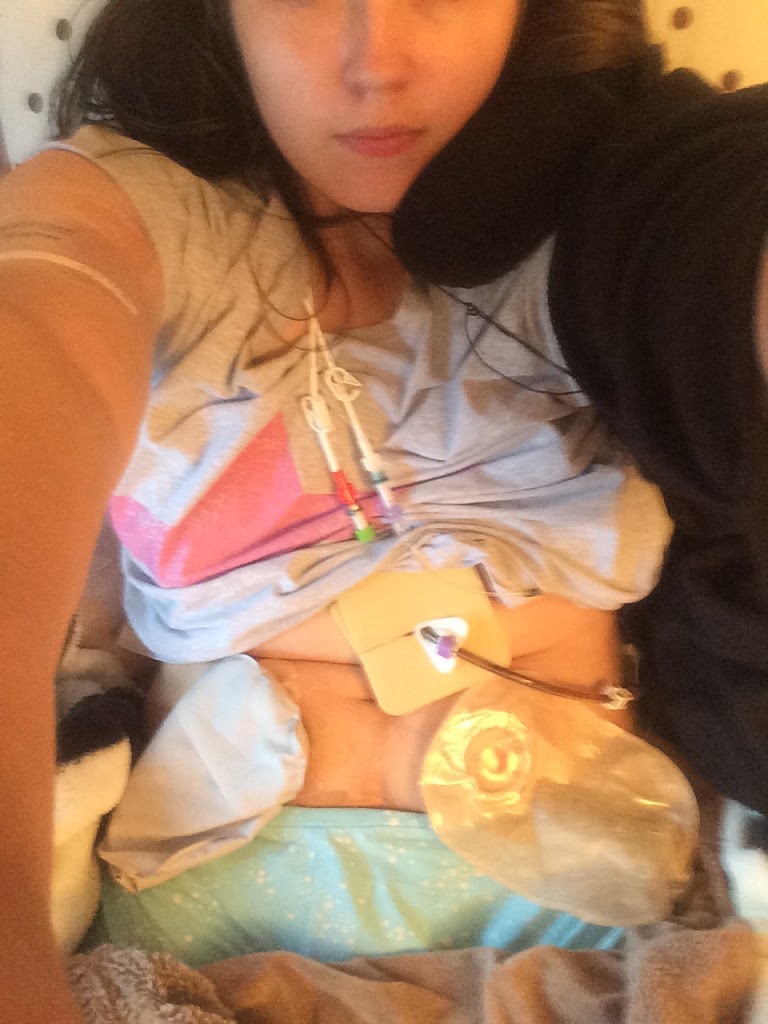
Knowing why I am the way I am and finally understanding why Ehlers-Danlos Syndrome didn’t cover all my symptoms and complications or the progressive nature of my condition, has been a huge relief. And the new ‘diagnosis’ of sorts has been agreed by all my specialists to make sense. They had their concerns but just referred to my condition as complex or atypical EDS. Now, it all makes sense, but it took this fresh, expert pair of eyes to comb through my life and see what others had missed all these years. I am 23 now, and it’s taken all that time to know what’s wrong, albeit I don’t yet have a diagnosis, we know what type of condition we’re dealing with. I will be undergoing a whole barrage of tests, including a muscle biopsy, as well as an inpatient stay, and I will be recruited onto the 100,000 Genomes Project to try and find the cause of my condition should the other tests not find the cause of my condition. I am desperate to know because I want my sister to be tested; I don’t want her future children to end up like me.
I am life-limited, my conditions will shorten my life expectancy considerably, but I try to make the most of every day. I am grateful for all I have, and that I live in a country with a free-at-the-point-of-use health service. I am very lucky. I am kept alive artificially by my TPN (feeding into the bloodstream) and the fluid and medication that gets pumped directly into my heart. I have expert interventions, a venting gastrostomy, an Ileostomy and a Urostomy. I have a care package, albeit far from perfect, that enables me to live at home, and I have a large team of specialists who fight to keep me alive and to give me the best quality of life that I can have.
Knowing I have a rare disease, and that the condition that has become a part of my identity, I might not have at all, has been a confusing new direction in my life. EDS is so ingrained in who I am, it’s almost like a badge of honour, but now I might not have it at all, and knowing, even if I do have EDS, it is not the cause of the majority of my complications, has taken a lot to get my head around. However, it’s a relief to know why I am the way I am. I still get to wear my zebra gear, but it represents a different condition now. My rare genetic or Mitochondrial Disease.

Today is rare disease day – do you care about rare? Please show your support if you can. Rare is everywhere, we just don’t realise. 1 in 17 of us will experience a rare disease at some point in our lives; it can happen to you. So please show you care about rare, and support all the people affected by a rare condition like me.
Thank you.
Related Posts



A very well written, well informed and informative article. Also, the uniqueness of the article is on point. So glad someone is talking about rare dieseases. Well done.