What if we viewed lived experience as an asset? As an asset of professional, economic,…
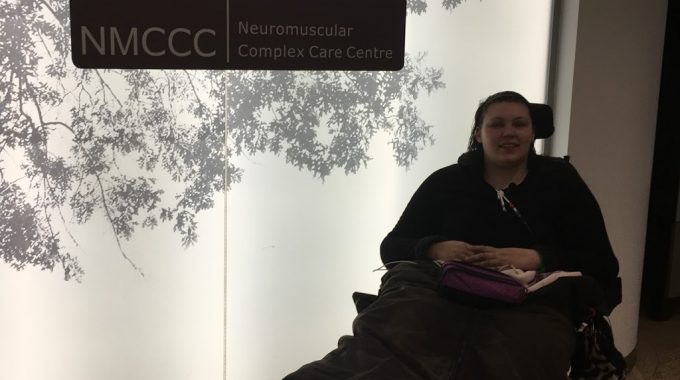
Hope For A Diagnosis: My NMCCC Admission March 2018 for a Muscle Biopsy

This week I was admitted to the Neuromuscular Complex Care Centre (NMCCC) at the National Hospital for Neurology and Neurosurgery, Queen Square (NHNN). This admission was for my muscle biopsy, long awaited – I’ve been waiting since 2016, and we had a last minute cancellation last year in July. I am cared for by Dr Ros Quinlivan, neuromuscular consultant who works across Great Ormond Street Hospital (GOSH) and the NHNN, and in charge of my care this admission was Dr Khan, who was with Ros’ team for this rotation. The staff on the NMCCC are fantastic too, mum stays with me to provide the 1:1 specialist care I require but they can never be more helpful and are so kind, you can always have a good chat and a laugh with the staff.
I was admitted on Wednesday, had bloods taken, respiratory function checked and so on, ready to have the biopsy. I found out I was on the list for Friday. So that was great, we were just waiting for that to happen. However on Thursday, when the surgical list was checked, I’d been removed from the list – no one knows why – by the administration team in the surgical department and then they also insisted that a form that had already been filled in and sent to them, hadn’t been done. Who knows why I was removed from the list, but there you go. So, the biopsy looked unlikely. Dr Khan worked extremely hard, phoning, emailing and so on to try and ensure I was put back on the list and had a slot to have my muscle biopsy, especially in light of how long I’ve been waiting and having had one cancellation already. He came to see me and told me it was looking unlikely, but that he’d keep trying and keep fighting. Dr Khan said he couldn’t promise me anything would come of it, but he promised to keep working on it – and he did. The best news was, he got my back on the list! So my biopsy could go ahead Friday. Late Thursday evening I was consented by the anaesthetist and we went to sleep in the knowledge that, barring any disasters/emergencies, my biopsy would go ahead.
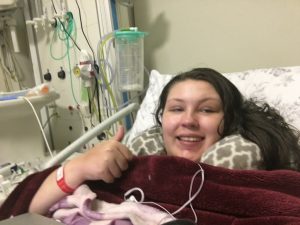
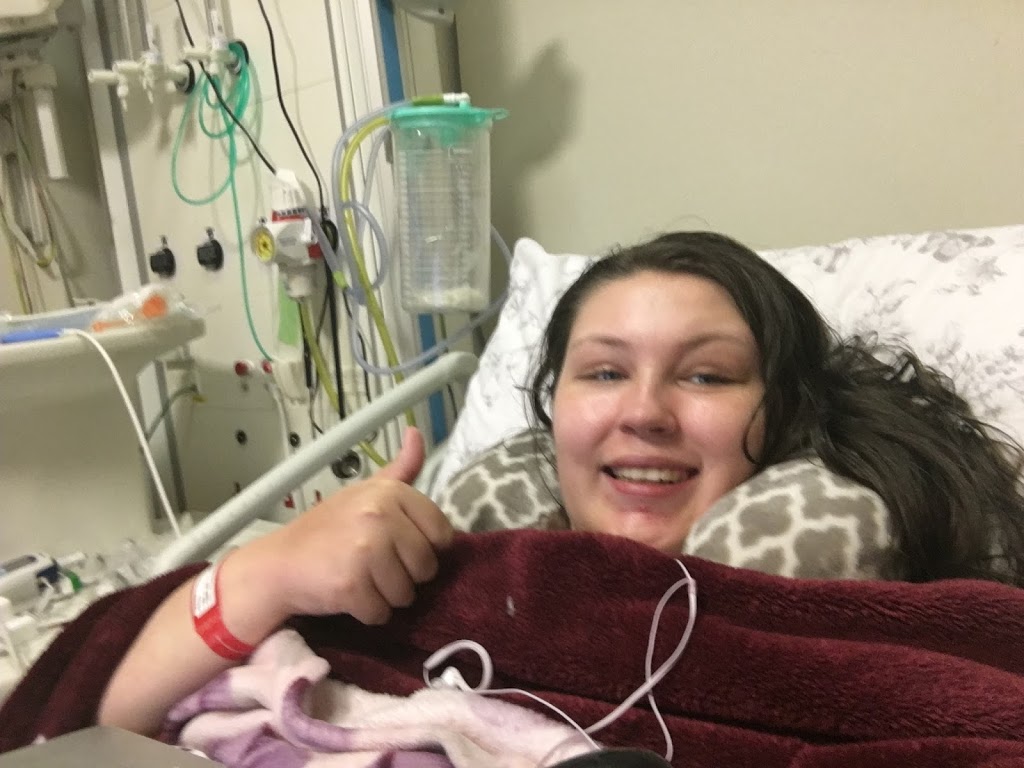
On Friday afternoon about 3pm, my biopsy went ahead. It was painful, I won’t lie. They can only numb the skin with local anaesthetic (I had it done awake), so when they get down to the fascia and then the muscle, there’s no anaesthetic they can put there, so you feel the whole thing, then digging around and cutting samples out of your muscle. They did it in my left thigh. It was very painful, the biopsies being taken, as was them cauterising the bleeding tissues afterwards, it really was painful, but not the worst thing I’ve experienced. It only took about 30 mins. I came back to the ward and then was allowed home today (Saturday). My leg is very very painful, more so than I was expecting and I’m usually so good with pain, but this I am struggling with. I just hope it eases up, as I have a busy week, including a meeting in London Tuesday and an appointment in London Thursday.


The muscle biopsy results could take 6 months or more, but some get their results back in less time. I just hope the biopsy might lead to a diagnosis, which it could do but there’s no guarantee and a biopsy doesn’t lead to a diagnosis for many, or I hope it’ll show features and defects that will guide us to know what kind of condition we’re looking at. Anything that might help with the diagnostic process.
People have asked why I am having a muscle biopsy and what we hope it’ll achieve. I hope this explanation will make it easier to understand. This is taken from a Facebook post I wrote before this admission:
“As you all know I was born with health problems that went undiagnosed – despite having lots of issues affecting lots of organs and systems and despite seeing many professionals through my childhood – until I became disabled and seriously ill at 14, then was diagnosed with hypermobile Ehlers-Danlos Syndrome a few days after my 15th birthday. Well, things just didn’t add up, EDS isn’t a progressive condition (whilst it can get worse over time, the condition isn’t by its nature progressive, from what I’m told and the literature that’s out there) and doesn’t often affect people in the way and extent it has for me and doesn’t usually shorten lifespans (in hEDS), whilst I recognise it does for some, it just didn’t add up for me. My team admitted it (calling it ‘atypical’ or ‘complex’ EDS) and everyone admitted there was probably something else going on, but no one seemed to do anything – I don’t think they knew what to do, who to refer to, where to send me. That was, until a registrar brought up mitochondrial disease in a routine appointment and my consultant referred me to the NHS National Rare Mitochondrial Disease Service, based at the NHNN, where I am seen by the incredible Dr Ros Quinlivan. Ros is child and adult trained and so treats children at GOSH and young adults and adults at the NHNN, hence naturally she took over my care when I was referred to the clinic. I saw Ros in 2016 and she ordered a muscle biopsy, a load of tests and wanted to admit me for my first annual “MOT”, which took place in July 2017. However the muscle biopsy got cancelled. I had an EMG which showed myopathy and then in the July admission I was found to have reduced lung function and capacity, and a weak/ineffective cough, so I was put on saline nebulisers to help loosen secretions and then a Lung Volume Recruitment (LVR – like a manual cough assist that uses breath stacking) to clear my chest, which has worked as I’ve had no chest infections since starting this regime. I saw a range of different professionals and fortunately my heart is okay at the moment, I was saturating okay with elevated CO2 at night but not high enough to warrant any ventilation (yay) so that was good, and lots of other tests and things. I had my bloods taken to have my genome sequenced by the 100,000 Genomes Project which, in conjunction with the muscle biopsy, we hope will lead to a diagnosis. Ros thinks it could be a rare Mitochondrial Disease or another progressive muscle-wasting disease, her words being “probably rare, ultra rare or even unique”. So, tomorrow I am admitted and whilst inpatient this time I will have my long-awaited muscle biopsy. The results will take a minimum of around 6 months, but could take up to a year or more (*I’ve since been told they could be back sooner than that*). I really hope we get a diagnosis, a name/a gene, even though getting a diagnosis won’t change my treatment, we know that. We may never get a diagnosis, but at least we’re trying with the fantastic help of Ros, the NMCCC and their teams.
However, whilst I want a diagnosis for myself, I also want a diagnosis so that my sister, Vicky is who is 27 (28 in July) can be tested to see if she is a carrier of the condition and if so, be counselled on the likelihood of passing the condition or carrying the condition onto her children and even have IVF with Pre-implantation genetic diagnosis (PIGD) to screen the embryos for the mutation and only implant the embryo(s) without the mutation, preventing the inheritance of the condition, thus meaning my sister’s future child(ren) would be unaffected. This is a huge driving force for me. I don’t want my sister to have children like me, and to go through what mum’s gone through. Vicky has suffered enough as a result of my illness, it affects her deeply and means she feels left out as mum isn’t able to devote the unlimited time to her as she has to be on-call and available to help me – even with the 24 hour care, which isn’t enough, as I need 2:1 care during the day but mum has to be on call regardless – so Vicky doesn’t get the time with mum that she needs, wants and deserves. She also often gets forgotten – people ask after mum and me but don’t always ask about Vic. She has suffered so much and I recognise that, it is hard for her. So, much as my illness and my life has taken a lot away from Vicky, I hope this gift of a diagnosis and a gene – if possible – that she can be counselled, tested and take measures to prevent her children inheriting it, that will be my gift and my legacy to Vicky. I can’t give her what she needs, I can’t change the situation, I can’t make things right, but I hope I can somehow make the future better for her by *potentially* preventing my illness being passed on, if Vicky was a carrier for the mutation.”
There’s a big change the biopsy won’t lead to answers, it could do but it might not. I just hope it might aid in the diagnosis in some way.
I am just so glad the biopsy is over and done with. Here’s to moving onwards with the diagnostic process. I am already on the 100,000 Genomes Project to have my genome sequenced, which may also lead to a diagnosis, but may not. I just hope beyond hope I will eventually know what is wrong with me. It’s so hard when you’ve spent a childhood with undiagnosed problems, to get a diagnosis at the age of 15, to then find after 8 years that what you thought was wrong with you, likely wasn’t, and then spend 2 years (thus far) not knowing what is wrong with you again. It’s hard not having a name or a cause, knowing there’s something wrong but no one knowing what. I do hope that I may get a diagnosis – even if it is rare, ultra rare or unique as Ros said, so much so it may not have a name – it’d just be good to know and possibly be able to take steps to ensure Vicky’s future child(ren) don’t inherit the condition. Fingers crossed!
Here’s my thank you to the NMCCC, Dr Quinlivan and Dr Khan.

Related Posts



This Post Has 0 Comments