What if we viewed lived experience as an asset? As an asset of professional, economic,…
Ehlers-Danlos Syndrome: Are You Aware?
We’ve all heard of cancer; the dreaded “C”. While there is still much to learn about cancer, and many lives lost to or affected by it, there are lots of charities to support these people and their families and a lot of money goes into researching it. I’m not detracting from this, or the horrendous treatments used to manage it, having had family members and friends affected by cancer, but I wish the same could be said for Ehlers-Danlos Syndrome. Lots of people are hypermobile, or so-called “double jointed”, which is what EDS is known for along with stretchy skin. However, not every “double jointed” person has EDS. EDS is so much more than that. Ehlers-Danlos Syndrome is a group of connective tissue disorders caused by a mutation of one or more of the genes that code for the production of collagen. It is hereditary, but some people, like myself, have what is known as a spontaneous mutation. This means there is no family history of the condition, we’re just very unlucky that the gene mutated in the sex cells or in our cells during our development. Think winning the lottery, but you get this debilitating, multi-systemic condition instead.
The mutation in the gene/s means our collagen is faulty. Think of collagen as the glue of the body. Your glue helps hold everything together, keeping your joints in the correct position, keeping your muscles strong, holds your organs together and allows your skin to stretch (up to a point) as you grow or put on/lose weight. For people with EDS, our collagen is more like over-chewed chewing gum. Instead of holding everything together, it stretches easily, is fragile and prone to tearing, doesn’t hold it’s shape and once stretched, it doesn’t go back. This means our ligaments stretch, leading to hypermobility, dislocations and easily torn ligaments; low muscle tone; our organs can stretch, collapse and even rupture; our blood vessels can bulge (aneurisms) and can be prone to rupture; our heart valves may be floppy and prolapse; our body struggles to hold itself together, meaning we have poor balance and our muscles have to work twice as hard to do basic things as other people’s; we have poor spacial awareness and knowledge of where our limbs are in relation to everything else (poor proprioception), making us seem very clumsy; gut and bladder problems due to stretching, weak muscles and problems with the nerves; organ prolapse; our necks can struggle under the weight of our heads (Craniocervical Instability); our spinal cord can be unusually tight causing Tethered Cord Syndrome and Chiari Malformation (where the brain herniates into the spinal canal); our Autonomic Nervous Systems don’t work properly (the ANS controls all the unconscious functions in the body: heart rate, blood pressure, temperature regulation, digestion etc.); we tend to develop osteoarthritis younger and at a faster rate; and much, much more. As much as 80% of the body is made up of collagen – hence why those with Ehlers-Danlos suffer so many multi-systemic problems.
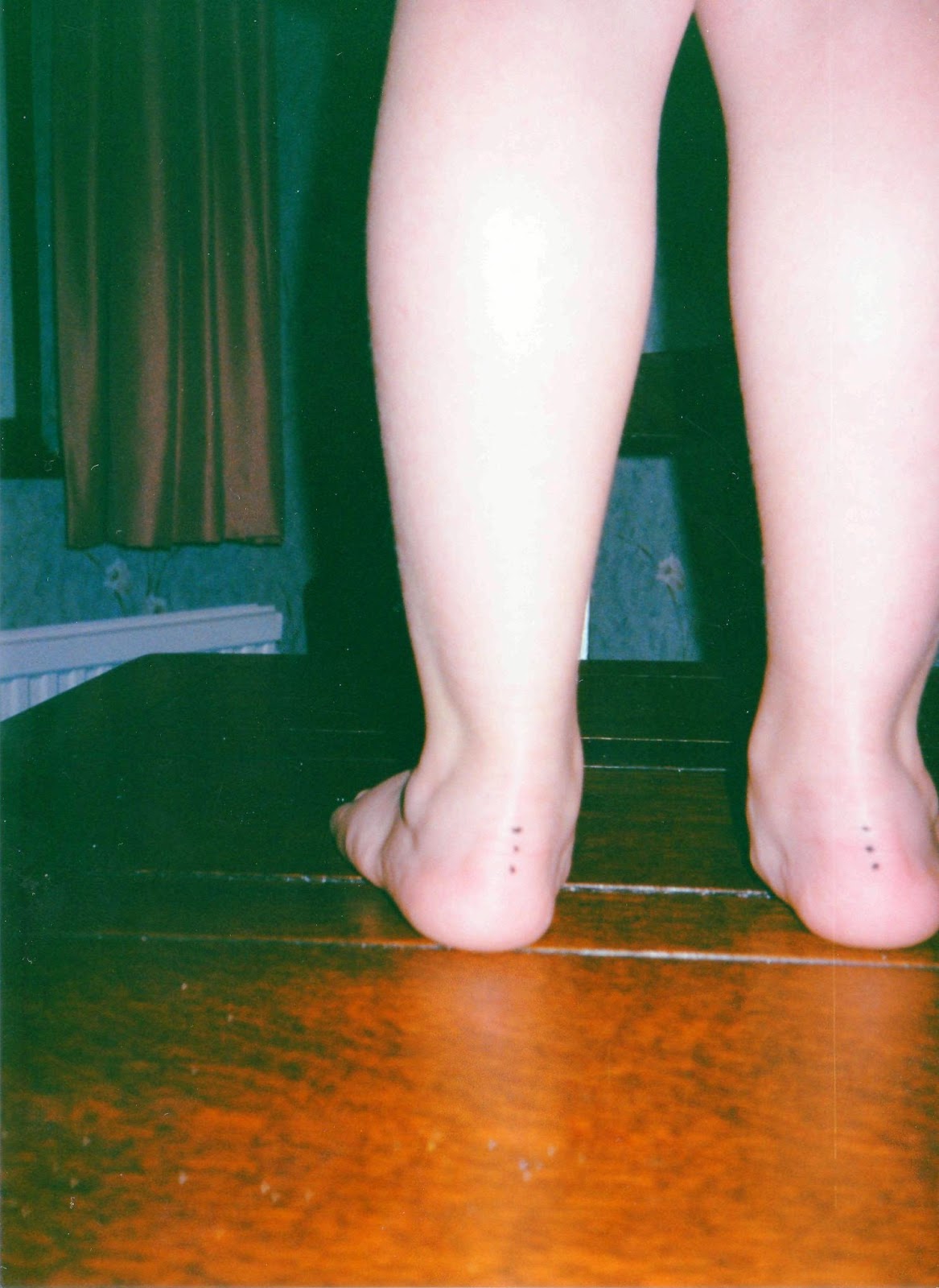 |
| Flat feet aged 5 |
Take me. I was born with terrible reflux that would make me stop breathing and turn blue if laid down, and I also suffered with anal fissures and constipation. I had hip problems (probably a dislocated hip) which led to septic arthritis at 18 months old and I was nearly put in a spica cast. Weak eye muscles and astigmatism requiring glasses. Finger deformities. Flat feet requiring orthotic insoles. I met my milestones late and wouldn’t put any weight through my legs and hated walking. However, I managed to have a somewhat normal life until I was 14, aside from having regular physiotherapy. In January 2008 I became disabled and my condition quickly progressed from there. First, my joints started dislocating, I was in chronic pain and began to suffer from fatigue. I put on a lot of weight due to inactivity and eating the wrong things. I became a wheelchair user. I was diagnosed with EDS a few days after my 15th birthday, and by my 16th birthday I couldn’t eat, couldn’t swallow, had tummy pain, felt full all the time, suffered with constipation, my bladder stopped working and I lost 6 stone in 6 months. I was being fed through a tube; at first a nasogastric “NG” tube, which goes up the nose and then down into the stomach, and then I had a PEG (gastrostomy) tube put in a few weeks after my 16th birthday. I was put on a lot of drugs to try and get my bowels moving, but all failed. I also started having dizzy spells and couldn’t sit up, so I was bed bound. I was diagnosed with Postural Orthostatic Tachycardia Syndrome and whole gut dysmotility, and I started intermittent self catheterisation to empty my bladder. We managed to, slowly, get me to a point where I could swallow safely so I could drink little bits for pleasure, but it wasn’t tolerated either.
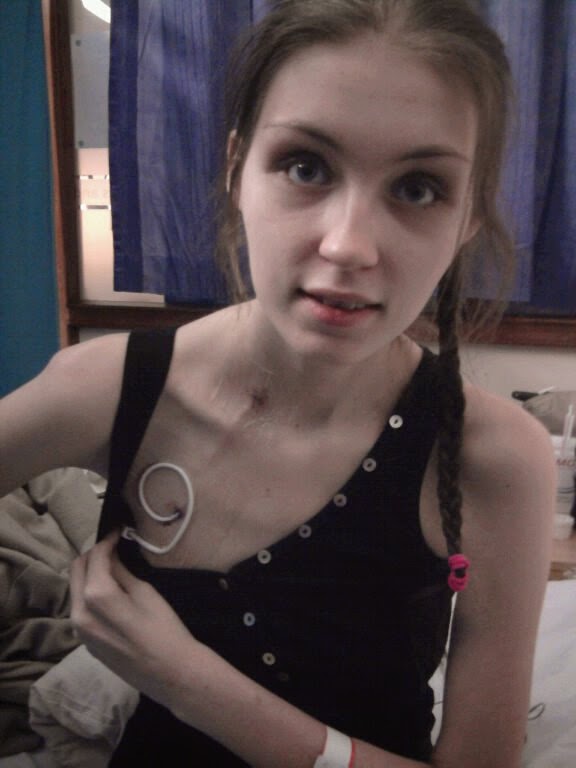 |
| The day my Hickman Line was put in |
Fast forward 18 months I’d lost 11.5 stones, I wasn’t tolerating or absorbing my feeds, we’d tried feeding me into my small bowel but that also failed, I was anaemic, covered in downy hair, looked awful (pictured) and now weighed 7 stone; very thin for someone of 6 ft 1. So on the 13th May 2011, after my hospice nurse frantically tried to save my life, I was admitted to hospital and put on Total Parenteral Nutrition “TPN”, which is feeding straight into the bloodstream. My reason for being on it was Pseudo Obstruction which had caused intestinal failure. It saved my life. It would have been days or weeks before I died had I not been put on TPN. I had my Hickman line (also known as a central line) put in on the 19th, which goes into my jugular vein and down into my heart, and the other end is tunnelled through my chest, coming out just above my right breast. The TPN is infused slowly through this line, which used to be overnight but now takes 21 hours to run through due to the volume.  Since then I’ve had an ileostomy (stoma bag) formed, a suprapubic catheter put in, diagnosis’ of spinal degeneration and Osteoporosis and am awaiting surgery for a Urostomy (stoma bag for urine), on the waiting list for spinal injections, will be having a heart monitor for a few days and will be having a small bowel MRI as my bowel is backing up and I have to have my stomach on to drain 24/7. I am mostly bed bound, I cannot walk at all, and have a wheelchair with a special recline function so I can lay myself down when my blood pressure drops. I have to have IV medication as I have no absorption, if not they have to be injected or dissolve and absorb through the gums (buccal). I am connected to my pump 24 hours a day between my TPN and IV medication. My mum has to do all of my IVs as I am not strong enough and my grip is too poor. I get terrible headaches, I live in severe pain, I sleep a lot now and my concentration is poor. I can only sit up for a few hours on a good day, which is extremely limiting. My quality of life is severely restricted.
Since then I’ve had an ileostomy (stoma bag) formed, a suprapubic catheter put in, diagnosis’ of spinal degeneration and Osteoporosis and am awaiting surgery for a Urostomy (stoma bag for urine), on the waiting list for spinal injections, will be having a heart monitor for a few days and will be having a small bowel MRI as my bowel is backing up and I have to have my stomach on to drain 24/7. I am mostly bed bound, I cannot walk at all, and have a wheelchair with a special recline function so I can lay myself down when my blood pressure drops. I have to have IV medication as I have no absorption, if not they have to be injected or dissolve and absorb through the gums (buccal). I am connected to my pump 24 hours a day between my TPN and IV medication. My mum has to do all of my IVs as I am not strong enough and my grip is too poor. I get terrible headaches, I live in severe pain, I sleep a lot now and my concentration is poor. I can only sit up for a few hours on a good day, which is extremely limiting. My quality of life is severely restricted.
All this in 6 years. And they say EDS isn’t progressive! This is my journey with EDS. What I haven’t explained is the disbelief, the people calling me fat and lazy, doctors denying EDS existed, various psychological therapies and assessments I’ve had, being told I had an eating disorder and being told it was all in my head, and I a school phobia (I am proud to say I loved school!). And much of this was after my diagnosis. It can take years to get a diagnosis; I am one of the lucky ones, it only took me 10 months. It needs to be recognised for how much it can affect people. No two EDSers (as we call ourselves) are the same. It’s not just hypermobility and stretchy skin!
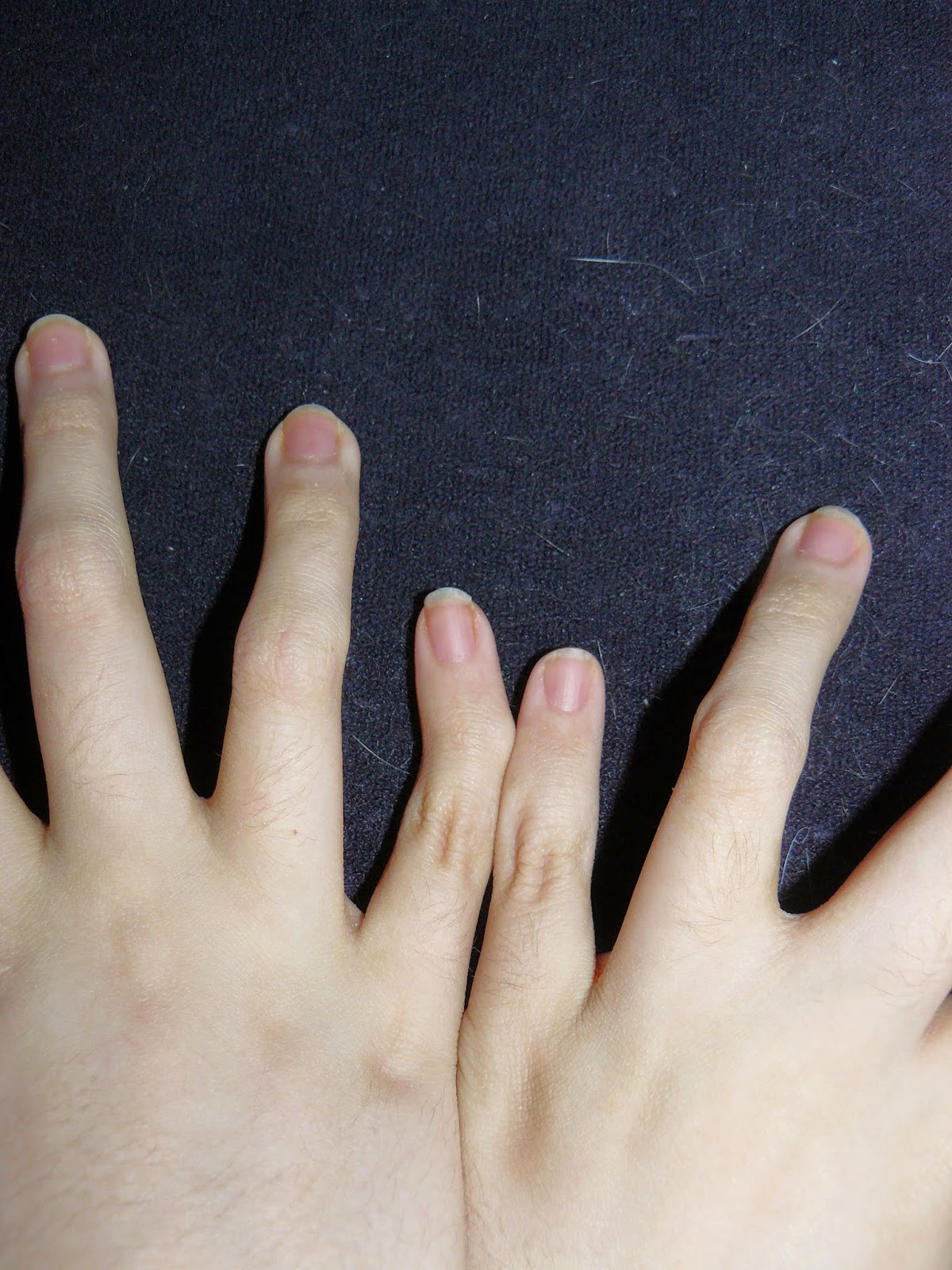
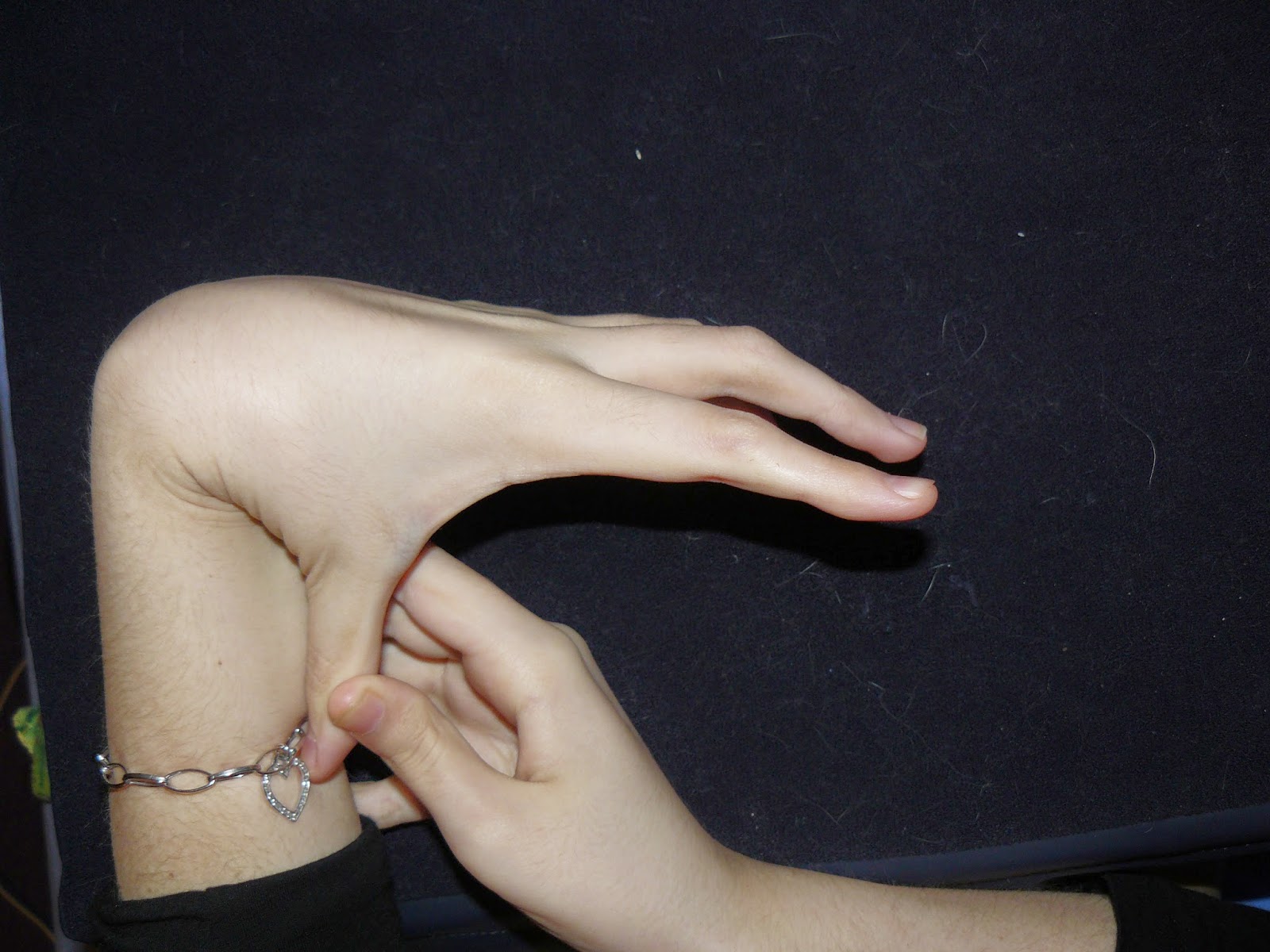
Ehlers-Danlos Syndrome can be life-threatening. The vascular type is fatal, with the average life expectancy being 48. The stretchy skin EDS is sometimes known for is most prevalent in the classical type. I have the hypermobility type, which to some is considered the “mild” type, but in reality it isn’t. H-EDS nearly killed me. Twice. The hypermobility type is more than just what its name suggests. This is a whole body condition; collagen is everywhere, bones, skin, organs, ligaments, muscles, blood vessels, teeth etc. There are a few more rare types, too. However, we often don’t conform to the problems known in each type, having crossovers between types. This just makes EDS even more confusing and difficult to diagnose.
May is Ehlers-Danlos Syndrome Awareness month. Read the Ehlers-Danlos Support UK website (here), google it, share this post; anything. Lets make our invisible, visible.

Here are a few photos showing how my EDS manifests itself
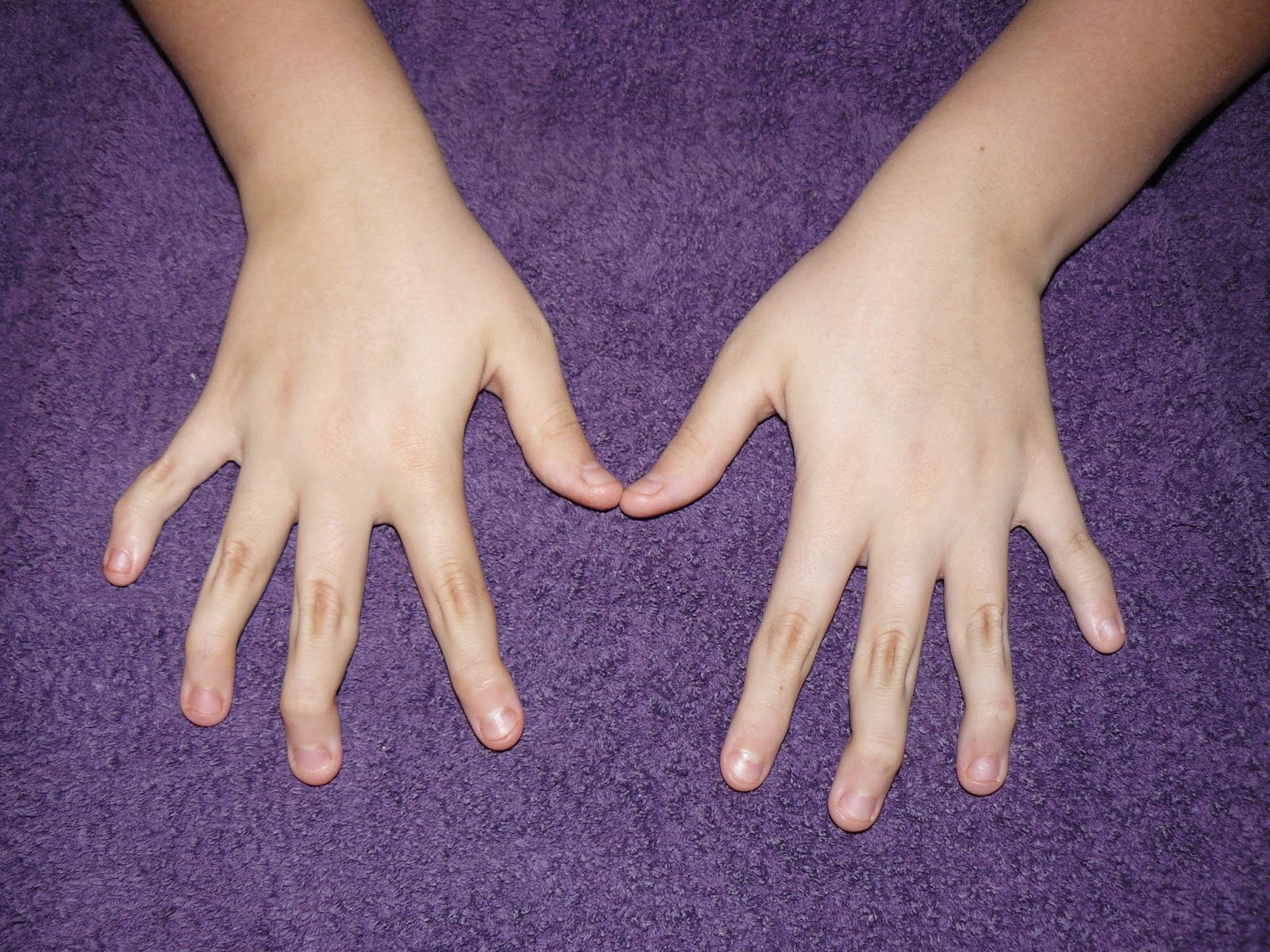




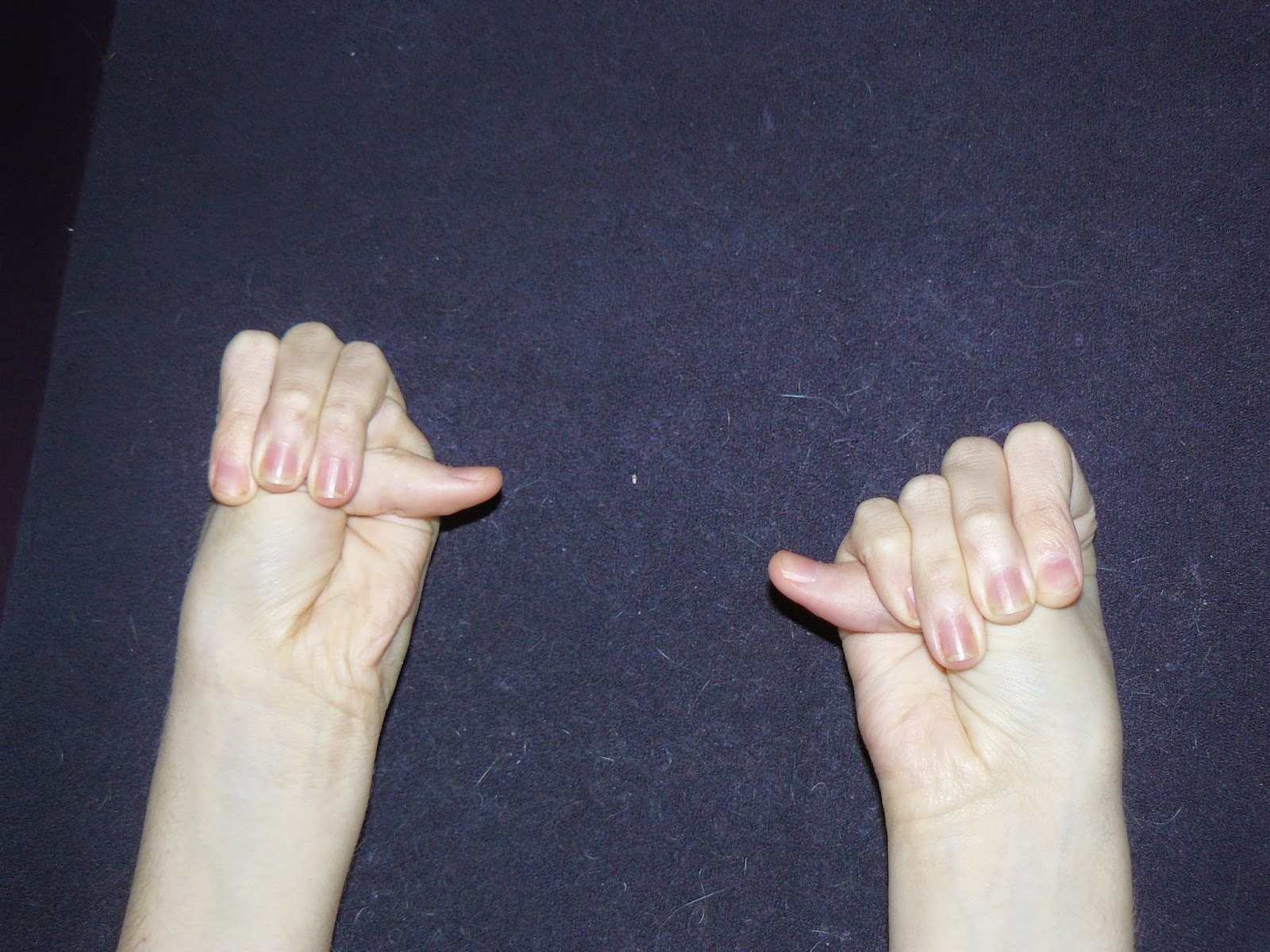




Related Posts



Amazing awareness article – I really didn't know much about EDS until I started talking to you on FB. The zebra image is good too. Many doctors think they will never see zebras!! Thanks for sharing so honestly. xx
Thank you for sharing and being so honest. My little has been diagnosed with hms and wears orthotics has had ddh and wears glasses along with heart mumur and asthma. I worry for her but no one listens really. Likewise my 14 yr old is show things that worry she keeps damaging muscles, tearing tendons and such like – all started in past 18 months.
You are brave beyond words. My heart goes out to you and your family. As a mother, I am sure your Mum despises everything this dreadful condition does to her baby. There are no words. 🙁
Lucy, with every word you write, people become more aware, more understanding. Keep writing. Beverley x
fantastic article and very bendy fingers! looks like you need ring splints hun ! if you do have a look at Ring splints by zomile on FB. x
Thank you Lucy for sharing your courageous story. My daughter Amy has this debilitating condition and her nutrition is totally dependant upon her peg gastrostomy. She has the pots and autonomic dysfunction too and diagnosis took 14 yrs. We all need to work together to raise as awareness of EDS, not just among the public but the medical profession too and that is exactly what you have just done! Well done x
You may want to look into nutrigenomics. HEDS is the only type of EDS for which the causative genetic mutation is unknown. Unlike the other types, for which mutations have been found, the incidence of Type 3 has absolutely skyrocketed in the past 15 years. There is no way this significant increase, within a single generation, could be attributed to a genetic cause. In my family, HEDS is epigenetic, meaning we have a genetic predisposition for disease that is then triggered by environmental exposures over the course of a lifetime. We can trace EDS back 4 generations. Our inborn joint hypermobility and neural tube defects were all the result of MTFHR deficiency in utero Like you, we were born with serious GI issues; severe constipation alternating w/explosive diarrhea, leading to anal fissures, chronic projectile vomiting, and failure to thrive requiring hospitalization. We went on to develop multiple neuro-immune/inflammatory/degenerative disorders – all complications from the underlying disease process going undetected and untreated for all those years. We were dx'd w/HEDS by a university rheumy, scored 9:9 on the Beighton. None of the geneticists here consider HEDS genetic in orgin. The second one we saw felt we also have the Classical type but did not order DNA testing as she said treatment would be the same regardless (only palliative) and also she was more concerned about our positive results for mito. Except for early onset arthritis and DJD/DDD, my EDS did not become disabling or painful until it turned inflammatory. At which point, my muscle tension became so tight that it started pushing/squeezing my hypermobile joints out of socket. I was having multiple full dislocations daily, and the skin ripping and spontaneous bleeds were increasing in frequency and severity. I literally could not move without ripping tissue. The geneticists concurred w/the super neurol and basically told us to go home and die, that there was nothing more they could do other than what we were already doing on our own. Geneticist #3 told us it would be another 10-15 years before the technology was available to isolate the mutation….typical allopathic cop-out! Had we listened, we would be dead. Instead, I took responsibility for my own healthcare. Within months, I regained partial use of my hands and the ability sit so was able to ramp up and complete my 29 years of research w/the discovery of the etiology of MTHFR in HEDS along w/the connection to dysautonomia and mast cell disease. In the process, I learned how to put most of our symptoms into remission – even my son's lifelong batwings have resolved – with a genetic-based nutrition program. I went from being too disabled to care for myself or even use a toilet back to my passion of sustainable organic farming. If I can do it, others can, too. The longer you wait to address cause, the harder it is to reverse. Here's a link to an article discussing my findings. I took the time to write it up and share this info free of charge in the spirit of helping others help themselves. As long as you do what you've always done, you'll continue to get the results you always got. If you need help, I'd suggest looking into Amy Yasko's protocol. She offers a step-by-step guide to recovery online free or charge. Best of wishes! http://www.mthfrheds.com
I am linking your post in my own EDS/chronic illness blog! Thank you for your wonderful post and your word illustrate strength. I am so glad for all the fellow zebras writing about their illness in the spirit of awareness. My Ehlers-Danlos has not progressed to this point as yours, so I can only relate on so many levels.
An excellent article. Thanks, I will share.
Not bad.
I have it too. Showed up two years ago, when I was 32, and just this week I am having trouble walking.
No one is going to learn from our suffering. Not when cancer patients get all the attention.
Anyway, I'm going to shoot myself. Fuck this sort of life. Ciao bella.
Thank you for your frank blog. I am currently in shock as my 7 year old son has been diagnosed with eds. Obviously I want and will do anything in my power to help keep him strong. I have been advised to get him on marine collagen which I will buy tomorrow from the specialist shop. I was told he will need to be on collagen for life. Marine collagen is expensive but I was advised I can pressure cook chicken bones (or other bones) to form a broth, to add to soup, casseroles etc. Do this 3 times a week, and he can get the collagen from that. Have you taken collagen supplements? Made a difference? I'm looking to get any support I can to help my son. If you have any advice that could help him, anything you've learned, please feel free to message me lcookee@rocketmail.com
I'm so sorry you have this affliction xxxxx
This comment has been removed by the author.
Lucy, you are an absolute inspiration. Your article made me cry but gave me a good insight into your life with EDS and a better understanding of what people who have sometimes hidden conditions must go through.
Your story resonates with me strongly. After lnowing i had hyper mobility for severall yesrs it has very suddenly crept up on me and complicated an earlier diagnosis of lupus. In the space of a few years i have gone from mobility and anolity to have a limited social and personal life to bring wheelchair bound with an ileostomy and SPC. I am in hospital currently after surgery to correct hernia only six months after ileostomy (history very like yours) complicated by emergency surgery for blockage and ischemic bowel …
I thought my story was inique however now i have gone online I have found others with stories similar to mine. Like lupus they can describe the problem but have nothing in the way of treatment or cure to offer us.
I can only say hang in there I hope it gets better for you perhaps soon someone will discover a way to treat EDS via genes?
My daughter has EDS. I am so pleased to see this because I have not seen much about this terrible condition, I feel so helpless. What can we do? Please